Video sobre sistemas do corpo humano:
https://youtu.be/eGks1VKcRQ0
terça-feira, 20 de novembro de 2018
domingo, 18 de novembro de 2018
Síndromes
O nosso corpo é formado por pequenas unidades chamadas células. Dentro de cada célula estão os cromossomos, que são responsáveis por todo o funcionamento da pessoa e suas características. Cada célula normal possui 46 cromossomos iguais, dois a dois, isto é, existem 23 pares de cromossomos, destes, 22 são chamados de cromossomos autossômicos e o outro par, chamado de cromossomos sexuais que são designados por letras, a mulher XX e o homem XY. Portanto, numa célula normal existem 46 XX cromossomos (mulher) ou 46 XY cromossomos (homem).
As desordens cromossômicas podem ser ocasionadas por migrações anômalas de cromossomos durante a formação do óvulo ou espermatozóide, ou ainda por alterações estruturais dos próprios cromossomos.
Algumas desordens cromossômicas causadoras da deficiência mental.
Aplicar-se-ão a esta condição os portadores da síndrome de Down com defeitos de discreta repercussão anátomo-funcional, e outras que por vezes apresentam grandes deformidades anatômicas do sistema nervoso e/ou físico-motoras. Em presença de distúrbio e a fim de melhorar a qualidade da vida de crianças atingidas por alterações genéticas acentuadas, como se sucede, por exemplo, na síndrome de Edwards (trissomia 13), ou na de Patau (trissomia 18), nas quais sabidamente a longevidade não ultrapassa o 1o ano de vida, buscam-se alternativas terapêuticas, como a musicoterapia e a equinoterapia.
As características principais das doenças de origem genética são: atraso mental, atraso do crescimento e, por vezes, malformação grave do coração. O crânio é excessivamente alongado ou alargado ou mesmo achatado, na região occipital e o pavilhão das orelhas apresenta poucos sulcos. A boca é pequena e o pescoço normalmente é muito curto. Há outras peculiaridades e uma infinidade de síndromes. Aqui estão descritas apenas algumas delas: Patau, Down, Edwards, Willians, Cri Du Chat, Marfan, Dandy Walker, Miller Dieker.
SINDROMES
Síndrome de Alport:
A síndrome de Alport é uma doença genética caracterizada por provocar a perda progressiva da função renal e auditiva.Também pode afectar o sistema visual. A presença de sangue na urina (hematúria) é quase sempre encontrada nesta condição.Foi identificada pela primeira vez numa família inglesa, por Cecil Alport, em 1927.
Causas:
Esta síndrome é causada por mutações nos genes COL4A3, COL4A4 e COL4A5, responsáveis pela síntese do colagénio. Mutações em qualquer destes genes impedem que a rede de colagénio tipo IV seja produzida. As membranas basais são finas estruturas laminares que separam e suportam as células. Quando mutações previnem a formação das fibras de colagénio tipo IV, as membranas basais das células renais não são capazes de filtrar correctamente o sangue, permitindo que o sangue e proteínas passem para a urina.
A síndrome de Alport pode ter diferentes padrões de hereditariedade dependentes do tipo de mutações genéticas.
Na maior parte dos portadores desta síndrome, a condição é herdada como ligada ao cromossoma X, devido a mutações no gene COL4A5. Uma condição denomina-se ligada ao cromossoma X quando o gene envolvido na desordem está localizado no cromossoma X. Nos indivíduos do sexo masculino, que têm apenas um cromossoma X, uma cópia alterada deste gene é suficiente para causar uma síndrome de Alport severa, explicando desta maneira a eventualidade de quase todos os indivíduos deste sexo desenvolverem insuficiência renal. Nos indivíduos do sexo feminino, que possuem duas cópias do cromossoma X, uma mutação numa cópia do gene COL4A5 resulta somente no aparecimento de sangue na urina, não havendo o desenvolvimento de insuficiência renal. Por ser um tipo de hereditariedade ligada ao cromossoma X, um pai não passará esta síndrome aos filhos do sexo masculino.
A síndrome de Alport também pode ser herdada de uma forma autossómica recessiva se as duas cópias do gene COL4A3 ou do gene COL4A4, localizados no cromossoma 2, sofrerem mutação. Muitas vezes, os pais de uma criança com uma doença genética autossómica recessiva não estão afectados por ela mas são portadores de uma cópia do gene alterado.
Síndrome de Brugada
A Síndrome de Brugada é uma arritmia hereditária (autossómica dominante) que provoca um rápido batimento dos ventrículos.É provocada por uma mutação no gene SCN5A, que leva a uma alteração estrutural dos canais de sódio do coração.Tem especial incidência em indivíduos adultos. Também ocorre com mais frequência em indivíduos de origem asiática.Pode provocar morte súbita (através de taquicardia ventricular polimórfica), principalmente em indivíduos em repouso ou durante o sono.
Síndrome de Cockayne
A síndrome de Cockayne é uma doença hereditária rara. Os pacientes afectados têm um excesso de sensibilidade à luz solar, têm estatura reduzida e sofrem de envelhecimento prematuro. O nome da síndrome advém do médico do Reino Unido chamado Edward Alfred Cockayne (1880 - 1956), primeiro a descrevê-la.
Síndrome de Crouzon
O Síndrome de Crouzon é uma doença genética conhecida como um síndrome do arco faríngeo, em especial do primeiro arco.
Síndrome de Dubin-Johnson
A Síndrome de Dubin-Johnson é uma doença recessiva autossômica que causa um aumento de bilirrubina sem elevação das enzimas do fígado (ALT, AST). Normalmente é diagnosticada na infância. O transporte de bilirrubina do fígado para o sistema biliar é anormal e a ela se acumula no fígado. Os pacientes possuem icterícia de baixo grau durante toda a vida, a qual pode ser agravada pelo consumo de álcool, gravidez, infecção e outros fatores ambientais.
Síndrome de Ehlers-Danlos
A síndrome de Ehlers-Danlos ou Cutis elastica é uma doença genética, do tipo autossómico. A sua incidência global é de 1 para 5000 nados-vivos. Os tipos 1 e 2 desta doença são os mais frequentes na população. É um defeito heredfitário de causas distintas. Pode ser um defeito na atividade do procolágeno peptidase na remoção das extremidades não-helicoidais do procolágeno, resultando na formação de foibrilas colágenas defeituosas. como tambpem uam mutação do gene que codifica a enzima lisil-hidroxilase, necessária para a modificação pós-transacional da lisina em hidroxilisina, resultando na diminuição da resistência da molécula de colágeno na síndrome.
Síndrome de Kelley-Seegmiller
Síndrome de Kelley-Seegmiller é uma desordem genética rara caracterizada pela formação de pedras na área urinária, começa como gota e sintomas neurológicos moderados. É causado por uma deficiência parcial da transferase de fosforibosil de hipoxantina-guanina.
Sintomas:
- Artrite gotosa
- Urolitiase
- Aumento de ácido úrico no sangue
- Cristalúria
- Urolitiase renal
Síndrome de Waardenburg
A Síndrome de Waardenburg é uma doença hereditária que se carateriza essencialmente por:
- Perda de audição.
- Mudanças na coloração do cabelo e da pele.
O primeiro a descrever esta doença foi o oftalmologista holandês Petrus Johannes Waardenburg.
Tipos:
- Tipo I: associado a mutações no gene PAX3
- Tipo IIa: associado a mutações no gene MITF
- Tipo IIb: associado ao locus WS2B
- Tipo IIc: associado ao locus WS2C
- Tipo IId: associado a delecção no gene SNAI2 gene. Muito raro.
- Tipo III: associado a mutações no gene PAX3
- Tipo IV: associado a mutações no gene EDNRB
Síndrome de von Hippel-Lindau
A síndrome de Von Hippel Lindau ou VHL é uma angioblastomose cerebelorretiniana, autossômica dominante com 100% de penetrância. É caracterizada pela presença de hemangioblastomas e carcinoma renal (carcinoma renal de células claras), anormalidades adrenais, pancreáticas e escrotais. Afeta igualmente homens e mulheres. Início da doença na 2 e 3 década. A Síndrome de von Hippel-Lindau é uma doença genética rara que envolve o crescimento anormal de tumores em partes do corpo particularmente irrigadas por sangue.
Características:
- Angiomatoses
- Hemangioblastomas
- Feocromocitomas - tumores na medula adrenal
Síndrome de Wiskott-Aldrich
A Síndrome de Wiskott-Aldrich caracteriza-se por desencadear uma imunodeficiência infantil ligada ao cromossomo X, sendo manifestada exclusivamente em meninos.
Origem no nome:
Dr. Wiskott descobriu os sintomas em 3 meninos da mesma família e o Dr. Aldrich a sua relação com o cromossomo X.
Sintomas:
A criança apresenta infecções constantes e de repetição (otite, pneumonia...), plaquetopenia (baixa contagem de plaquetas), eczemas na pele (grosseirões, manchas de tom púrpura...) e sangramentos espontâneos (principalmente nasais e gengivares).
Diagnostico:
Exame de DNA em amostra sanguínea da mãe e do filho confirmando o traço genético.
Tratamento:
Transplante de medula óssea (o mais recente e com mais resultados é o de cordão umbilical).Quanto mais cedo o transplante, maiores as chances de cura.
Síndrome de Liddle
A síndrome de Liddle é um desordem autossômica dominante que imita o hiperaldosteronismo. Envolve problemas com ressorção de excesso de sódio e perda de potássio do túbulo renal. A hipertensão começa cedo, freqüentemente em infância.
Síndrome de Marfan
A síndrome de Marfan, também conhecida como Aracnodactilia, é uma desordem do tecido conjuntivo caracterizada por membros anormalmente longos. A doença também afeta outras estruturas do corpo, incluindo o esqueleto, os pulmões, os olhos, o coração e os vasos sangüíneos, mas de maneira menos óbvia. Seu nome vem de Antoine Marfan, o pediatra francês que primeiro a descreveu, em 1896. A síndrome de Marfan é uma doença genética associada a deficiências do tecido conjuntivo (desempenha uma função de suporte nos diversos órgãos do corpo). Como resultado, os indivíduos com esta doença apresentam frequentemente anomalias a nível esquelético, ocular e cardiovascular, entre outras. Muitos dos indivíduos afectados têm alterações das válvulas cardíacas e dilatação da aorta. As complicações cardiovasculares mais importantes em termos de risco de vida são os aneurismas da aorta e as dissecções da aorta. A prevealência é de aproximadamente 1 em 5000 indivíduos.
Síndrome de Miller-Dieker
A síndrome de Miller-Dieker caracteriza-se por apresentar uma lisencefalia tipo I, associada a dismorfias faciais como afundamento bitemporal e mandíbula pequena, se descreve uma mutação no cromossomo 17.
Síndrome de Sanfilippo
A Síndrome de Sanfilippo se apresenta com quatro tipos (A, B, C e D) que envolvem enzimas diferentes entre elas Heparan N-sulfatase(tipo A) no tipo D o gene foi mapeado no 12 q14. Inicia-se entre 2-4 anos e geralmente morrem na puberdade. Pode ser detectada em exame das vilosidades coriônicas
Sintomas:
- Retardo mental progressivo, inclusive marcha, fala e comportamento;
- Hiperatividade;
- Hepatoesplenomegalia variável ;
- Severo retardamento psicomotor;
Síndrome de Stickler
A síndrome de Stickler (também denominada artro-oftalmopatia hereditária, síndrome de Marshall-Stickler ou síndrome de Wagner-Stickler) é uma doença genética. Tem uma transmissão autossómica dominante, com expressão variável. É provocada por uma mutação que ocorre num gene associado à produção do colagénio.A ocorrência na população é de 1 para 20000 pessoas.
Alguns dos sinais diagnosticantes são:
- Fissura palatina (fenda palatina)
- Face plana
- Miopia intensa (com descolamento da retina e ocorrência de cataratas.
- Deficiências auditivas
- Artropatia (com displasia espondiloepifisária).
- Surdez
- Astigmatismo
- Artrose
- Laxidez articular
- Aracnodactilia
- Micrognatia/retrognatia
- Anodontia/oligodontia
- Cifose
- Glaucoma/buftalmia
- Alterações do esmalte dos dentes
- Amiotrofia/agenesia muscular
- Erupção prematura dos dentes
- Membros longos
- Hipotonia
Síndrome de Usher
A síndrome de Usher é uma doença genética que causa surdez e cegueira. É basicamente uma retinite pigmentosa de carácter progressivo combinada com deficiências auditivas graves de natureza congénita. É normalmente uma doença autossómica recessiva, afectando uma em cada dez mil pessoas.
Herança do sexo 1 e 2
herança 1- HERANÇA LIGADA AO X, HERANÇA LIMITADA PELO SEXO E HERANÇA INFLUENCIADA PELO SEXO. Os cromossomos sexuais não são completamente homólogos, e portanto devese esperar que os padrões de herança relacionados ao sexo do indivíduo sejam diferentes daqueles dos autossomos. Por exemplo, os genes presentes apenas no cromossomo sexual X serão representados duas vezes nas fêmeas e uma vez nos machos. Logo, espera-se que os recessivos desse tipo apareçam com mais freqüência no fenótipo de machos. Os genes localizados exclusivamente no cromossomo X são denominados genes ligados ao sexo ou genes ligados ao X. Já os genes que ocorrem apenas no cromossomo Y só produzem efeitos nos machos, sendo estes os genes holândricos. Conhecem-se ainda outros mecanismos pelos quais um dado caráter é limitado a um dos sexos (genes limitados ao sexo), ou mesmo em que a dominância de um alelo dependa do sexo do portador (genes influenciados pelo sexo). I) Herança ligada ao X: Quando o gene alterado está no cromossomo X. Este tipo de herança pode ser recessiva, ou seja, as manifestações vão estar presentes nos machos porque eles têm apenas um cromossomo X, ou seja, não têm nenhum gene normal para aquela característica e nas fêmeas quando existe alguma manifestação clínica em geral é mais leve. É exemplo desse tipo de herança a hemofilia. O risco de um filho ser afetado quando sua mãe carrega um gene alterado em um de seus cromossomos X é de 50% e das filhas é também 50% de serem portadoras do gene com defeito e consequentemente passar a seus filhos e filhas. É importante lembrar que nas aves o sexo homogamético é o masculino (ZZ) e o heterogamético, o feminino (ZW). Pode-se citar como exemplo de herança ligada ao sexo, a presença de barra larga na pelagem dos machos (BB). As fêmeas só podem apresentar barra estreita (B_) ou serem desprovidas de barra (b_), enquanto os machos podem ter barra larga (BB), barra estreita (Bb) ou ausência de barra (bb). Isso ocorre, porque o gene responsável pela barra localiza-se exclusivamente no cromossomo Z. Em humanos, conhecem-se mais de 150 caracteres confirmados ou altamente prováveis de ser ligados ao X. Dentre eles pode-se citar duas formas de diabete insípido, uma forma de displasia ectodérmica anidrótica (ausência de glândulas sudoríparas e de dentes), ausência de incisivos centrais, certas formas de surdez,
Herança 2- Nistagmo, cegueira noturna, atrofia óptica, glaucoma juvenil e distrofia muscular juvenil. Daltonismo: Um famoso exemplo é o daltonismo tipo verdevermelho (Daltonismo deutan), no qual faltam os cones células sensíveis à luz- sensíveis ao verde. As mulheres podem ser homozigotas normais, heterozigotas ou homozigotas raramente para o alelo defeituoso, pois possuem dois cromossomos X e, portanto, uma probabilidade maior de receber o alelo normal de ao menos um dos pais. As mulheres heterozigotas variam no grau em que a visão de cores é afetada, pois depende da proporção de suas células retinais que expressam o alelo normal. Já os homens recebem um alelo dominante (normal) ou um alelo recessivo (visão defeituosa para as cores vermelha e verde). Conhecem-se também outras formas de daltonismo, alguns ligados ao X e outros autossômicos. Existem testes para detectar daltonismo baseados em quadros formados por pontilhados. Neste teste, uma pessoa normal lê 74 e uma daltônica, 21. Hemofilia: Outro exemplo de condição recessiva ligada ao sexo é a hemofilia. Este distúrbio caracteriza-se pela coagulação do sangue deficiente devido à falta de determinadas proteínas que participam do processo de coagulação sangüínea. Como essas proteínas ou fatores de coagulação atuam em cadeia, a ausência de uma delas torna a coagulação muito lenta e o indivíduo tem tendência a hemorragias. Existem dois tipos de hemofilia ligada ao sexo: - Hemofilia A: caracterizada pela falta de globulina anti-hemofílica (Fator VIII), ocorrendo em 4/5 dos casos de hemofilia. Ela é conhecida nas famílias reais da Europa, em que pode ser acompanhada até a rainha Vitória, que deve ter sido heterozigota. A hemofilia em seus ancestrais não é relatada, supondo-se que o alelo para hemofilia surgiu de um gameta mutante. - Hemofilia B (ou Doença de Christmas): defeito no componente tromboplastínico do plasma (PTC ou Fator IX),
sendo mais drástica do que a hemofilia A. Ela também ocorre em cães.
quarta-feira, 14 de novembro de 2018
Sistema Rh
O sistema Rh foi descoberto em 1940 pelos cientistas Landsteiner e Wiener, usando hemácias de macacos da espécie Macaca rhesus, por isso o nome sistema Rh. Landsteiner e Wiener injetaram as hemácias desses macacos em coelhos e observaram que foram formados anticorpos para tentar combate-las. O sangue das cobaias, no caso os coelhos, foi centrifugado e pode-se obter um soro que continha os anticorpos que aglutinavam o sangue dos macacos. Esses experimentos trouxeram a descoberta do antígeno na membrana das hemácias que eram diferentes dos aglutinogênios A e B, ao qual chamaram de anti-Rh.
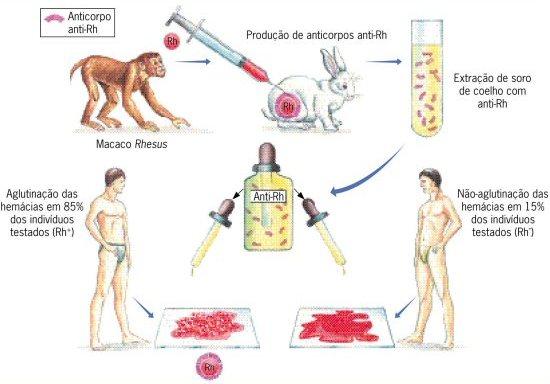
Esse antígeno é controlado por genes independentes, ou seja, os genes do sistema Rh não têm ligação com os genes do sistema ABO. Podemos encontrar as mesmas proteínas das hemácias em muitos animais diferentes, como por exemplo os humanos e os macacos superiores, que podem compartilhar vários tipos de sistema sanguíneo existentes. Isso infere em um indício evolutivo para essas espécies.
Na experiência realizada por Landsteiner e Wiener, gotas de sangue de um indivíduo humano que continha o soro anti-Rh, mais de 80% dos indivíduos apresentavam aglutinação e somente o restante não apresentava nenhuma aglutinação. Concluiu-se então que o grupo em que a amostra de sangue aglutinava apresentava o antígeno Rh e eram chamados de grupo Rh+ e o que não aglutinava não possuía o antígeno, sendo assim chamado de grupo Rh-.
Os indivíduos negativos são vão apresentar anticorpos se receberem, em algum momento de suas vidas, hemácias com Rh positivo. A herança do fator Rh é condicionada por três genes: RR, Rr ou rr, sendo R o alelo dominante que expressa o fator Rh+ e r o alelo recessivo que expressa o fator Rh-.
| Genótipos | Fenótipos |
| RR | Rh+ |
| Rr | Rh+ |
| rr | Rh- |
Eritroblastose fetal
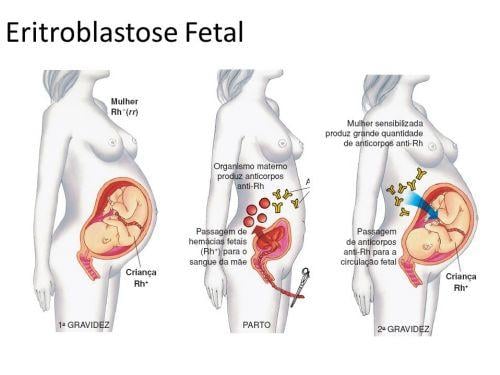
A diferença de fator Rh em um casal pode provocar a doença hemolítica do recém-nascido, conhecida também como eritroblastose fetal. Isso acontece quando uma mulher Rh- tem filhos com um homem Rh+ pois, nesse caso, existem duas possibilidades de fator Rh para os filhos, que serão condicionadas caso o homem seja puro (RR) ou híbrido (Rr). Caso o homem seja puro, todos os filhos desse casal serão Rh+, caso ele seja híbrido, podem nascer tanto filhos Rh+ quanto Rh-.
Quando o primeiro filho apresenta Rh-, ou seja, o mesmo da mãe, não existe incompatibilidade pois os dois não possuem antígenos. Entretanto, se o primeiro filho for Rh+ a mãe poderá entrar em contato com as hemácias do filho no parto e até mesmo dias antes do nascimento quando uma pequena quantidade de sangue do feto escapa para o organismo materno e, ser sensibilizada e passar a produzir o anticorpo anti-Rh.
A produção desse anticorpo não é imediata e o primeiro filho não terá incompatibilidade com a mãe, porém se esse casal tiver mais um filho com Rh+, durante a gestação os anticorpos da mãe já estarão concentrados no sangue e podem atravessar a placenta, provocando assim a aglutinação das hemácias do feto, ai então a criança será portadora da doença hemolítica do recém-nascido ou eritroblastose fetal, que pode causar a morte do bebê.
Em muitos casos graves, ocorre um aborto espontâneo quando a eritroblastose fetal acomete o feto. Se a criança nascer, ela pode ser salva se houver uma troca gradativa de seu sangue por outro que possua Rh-, dessa forma as hemácias não serão destruídas e o organismo da criança terá tempo para eliminar os anticorpos da mãe até que ele produza hemácias Rh+ novamente.
A eritroblastose fetal pode ser prevenida se logo após o primeiro parto de uma criança Rh+, a mãe Rh- receber uma aplicação de anticorpos anti-Rh. Eles destruirão as hemácias positivas que o feto deixou no sangue da mãe e impede a sensibilização do organismo dela, isto é, o desencadeamento da produção de anticorpos maternos que causarão problemas no segundo filho. Como o corpo da mãe não “aprendeu” a fabricar anticorpos, a mãe fica livre para ter outro filho sem a possibilidade da doença.
Sistema MN
Existem dezenas de sistemas sanguíneos utilizados na espécie humana pois podemos encontrar muitos antígenos diferentes na superfície das hemácias. No sistema MN os dois genes encontrados são conhecidos como LM e LN. O gene LM produz antígeno M e o gene LN produz antígeno N. Esses genes são codominantes e por isso esse grupo apresenta três genótipos e três fenótipos também, veja o quadro a seguir.
 Genótipos e fenótipos do sistema MN
Genótipos e fenótipos do sistema MN
Nesse sistema a produção de anticorpos também só acontece após a sensibilização. O estudo desses grupos sanguíneos diferentes é importante para determinar características de origem e evolução, para realizar transfusões sanguíneas e também para determinação de paternidade nas populações humanas.
Assinar:
Comentários (Atom)